
蛋白質純化
RP-HPLC 是一種有效的蛋白質/多肽純化工具。
通過 RP-HPLC 法可以從雜質中分離目標蛋白/多肽,采集到的片段可用于進一步研究,以及借助正交分析技術的分析,甚至可作為治療藥物。
在蛋白質/多肽分析過程中,色譜條件優化的目標是優化分辨率和保留時間。
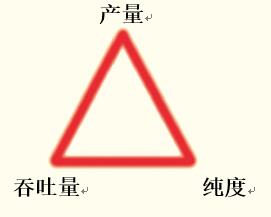
制備色譜法分離蛋白質/多肽時,色譜條件的開發主要是三個參數的優化(參見圖45):
- 產量是從色譜法每一步中得到的純化的目標蛋白/多肽含量。高產量可提高純化過程的實用性,并降低成本。
- 純度是從目標產物中去除雜質的程度。純度高有助于從后續分析中獲得更佳的數據或獲得高純度產物。
- 通量用來衡量制備周期中純化的物質量。高通量說明在給定的成本和時間內獲得更多研究或分析用物質,或更多原料藥,用于制藥領域。
由于制備色譜的目的與分析色譜的目的不同,因此色譜條件優化也不同。
圖45. 在蛋白質或多肽的制備純化中,通過尋求產量、純度及通量的最佳平衡實現分離條件的優化。
樣品裝載
在分析色譜中,將小樣品裝載到色譜柱上以確保加樣量不影響分辨率。
如果樣品量過高,則峰會加寬,進而分辨率會下降。
在不發生峰展寬的情況下允許加載到色譜柱的樣品量(“樣品容量”)取決于柱的大小(附錄列表顯示了柱的大小和樣品容量)。
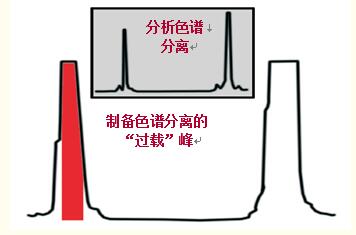
制備色譜法純化蛋白質/多肽時,通常會超出樣品容量,使柱“過載”,從而增加產量和通量(圖46)。
當允許一定的分辨率損失時,加載的樣品量可為樣品容量的10~50倍(附錄,最大實際負載)。
圖46中,盡管由于柱過載使得峰變寬,但峰形相對較好,說明嚴重過載在增加產量的同時還能保持純度,但目標蛋白/多肽的損失不可避免。
由于樣品過載,圖47中峰也同樣加寬。
片段收集、分析和利用
當柱過載時,通常會拋棄起始峰和結尾峰。
圖46中,對紅色區標示的峰的中間區域進行了收集。去掉了峰首和峰尾。
這避免了分離度較差的雜質峰的收集,增加了純度,但降低了產量。
在制備色譜中,對幾種感興趣的峰進行了片段收集,用于雜質分析。
圖46.在多肽純化示例中,制備分離包括柱的過載加樣,以增加產量。
分辨率降低,為了增加純度必須拋棄峰首和峰尾,但產量稍微有所降低。
基于分析結果,收集了少雜質或無雜質的片段,拋棄了峰首和峰尾附近雜質較多的片段。
收集和拋棄片段的選擇應考慮純度和產量的平衡。
例如,在不損失分辨率的情況下,4.6 x 250 mm的“分析”柱可用于純化少量多肽(最多約200微克)。
但為了增加產量和通量,同樣大小的柱最多可純化10毫克,但會有一定的純度或產量損失。
恰當的收集峰選擇有助于實現純度和產量間的最佳平衡。
在制備色譜中,盡管重點在樣品質量,但樣品體積也可能會很大。
盡管可采用樣品環和注射器,但通過將樣品“泵”至柱上可以注入更多樣品。
將泵的吸入管置于樣品容器內,樣品通過洗脫泵加載至色譜柱。
當有機溶劑濃度低(通常,樣品裝在水相中)且目的蛋白/多肽以有機溶劑梯度洗脫時,可通過這種方式裝入大量樣品。
吸附劑粒徑
分析色譜常用的吸附劑粒徑為5μm,制備色譜常用的吸附劑粒徑更大。
尤其是加樣量超過樣品容量時(柱過載),柱效較分析色譜影響較小。
當柱過載時,大粒徑填充柱同小粒徑填充柱分離蛋白質和多肽的效果一樣。
因此,制備色譜常用10μm或以上粒徑的吸附劑。粒徑分布也往往更寬。
不同于0.5μm或更窄的粒徑分布范圍,制備色譜的粒徑范圍更大,例如10~15μm。
由于制備色譜柱反壓和成本更低,因此更傾向于采用大粒子。
柱內徑
由于樣品容量很低,純化過程很少使用小孔柱(內徑小于2mm)。
小規模實驗室純化采用細孔柱(內徑約2mm)和分析柱(4.6 mm內徑)。
這種小規模制備分離的色譜條件通常與分析分離的色譜條件相同。
需要大量蛋白質/多肽時,采用10mm和22mm內徑的柱子。
1 mg蛋白質或多肽的純化可采用10 mm柱子,5 mg純化可采用22 mm柱子。
允許柱過載時,可純化更多蛋白質/多肽,10mm柱最多可純化50mg,22mm柱最多可純化200mg。
大量蛋白質或多肽的純化采用50 mm、100 mm或內徑更大的大內徑柱子。
50mm內徑的柱上已知最多可純化5克蛋白質/多肽。
柱長
與分析柱相比,制備柱往往相對較短。
這是因為在制備色譜法中,柱的總體積比柱長更重要,特別是蛋白質的分離。
內徑60cm、柱長12-15 cm(“圓餅狀”柱)的色譜柱已應用到蛋白質治療藥物的大規模純化中。
由于在制備色譜法中,柱通常過載,且效益的重要性遠不及產量、純度及通量重要,因此根據其實用性而非效益優化柱尺寸。
流動相組成
同分析色譜法一樣,采用10~22mm內徑柱的小規模純化常用乙腈-TFA體系。
大規模純化通常采用乙醇等溶劑替代乙腈,采用乙酸替代TFA。
盡管采用這些溶劑作流動相會降低分辨率,但它們更適合大規模使用,且分辨率的降低與柱過載固有的分辨率損失相同
蛋白質變性
通常認為反相色譜法會使蛋白質變性,因此洗脫出來的蛋白質不是天然蛋白,且可能不具備生物活性。
盡管反相HPLC的操作條件會使蛋白質變性,但洗脫后仍可獲得天然、具有生物活性的蛋白質。
有機溶劑可能減弱疏水力,造成蛋白質三級結構的損失。吸附劑的疏水面也可能導致蛋白質的去折疊。
但與色譜分離時間相比,蛋白質去折疊通常較慢,且蛋白質在反相色譜分離期間僅發生輕微變性。
由于二硫鍵的作用,蛋白質保持球形結構,且僅發生部分去折疊,因此從反相柱洗脫出的蛋白質通常可通過在恰當的重折疊緩沖液中處理,從而恢復其天然結構而恢復至天然狀態。
目前有許多實例均顯示采用反相HPLC純化后的蛋白仍維持天然三級結構和生物活性。
胰蛋白酶的反相純化,其中活性得到了保留,且隨后用于蛋白質的胰蛋白酶酶切。
重組人紅細胞生成素是一種成功商業化的蛋白質治療藥物,采用了反相高效液相色譜法將蛋白藥物從其細胞培養表達系統中分離出來。
采用反相HPLC對另一種商業蛋白質治療藥物——粒細胞刺激因子進行了純化。
此外,還采用反相HPLC對重組人胰島素進行了純化,維持了活性結構。
純化實例
圖47顯示了合成肽——促性腺激素釋放激素(GnRH)拮抗劑的純化過程。該純化過程通過以下幾個步驟展開:
- 在4.6 x 250 mm 的分析柱上構建洗脫條件。
- 在50 x 300 mm 的柱上裝載1.2克合成肽混合物,基于第一步構建的條件洗脫(圖47)。
- 對 GnRH 拮抗劑各洗脫峰的片段進行收集并借助分析法分析,最終實現最高產量和純度。
- 用乙腈和TFA作為洗脫劑,在反相色譜柱上再次進行色譜分析,完成收集到片段的脫鹽處理。
-
收集片段實現最高產量和純度。
在該色譜純化步驟中,從1.2 gm的反應混合液中可收集128 mg的純化肽。
該純化過程采用了與分析色譜分離相同的有機溶劑——乙腈,但采用磷酸三乙胺替代了TFA。
由于柱過載,洗脫峰更寬,分辨率不如分析色譜。但峰仍然較為緊密,且容易收集到含目的多肽和GnRH的洗脫液。
圖47. 128mg的合成肽——促性腺激素釋放激素的純化。
柱嚴重過載,導致峰非常寬。
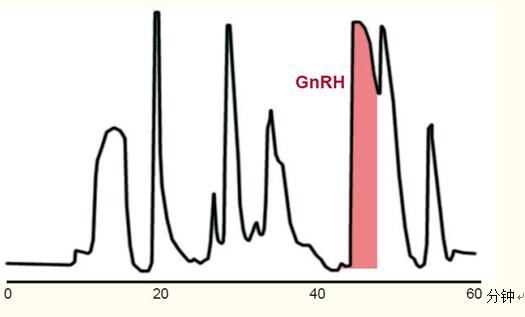
條件
色譜柱:C18寬孔柱,15~20μm粒徑,50 x 300 mm。
流動相:乙腈和磷酸三乙胺水溶液梯度洗脫。
樣品:促性腺激素釋放激素

